
Jean-Marc JOUBERT – ICMPE, UMR CNRS 7182, Université Paris-Est Créteil
Introduction
Ce tutoriel montre, sur un exemple concret, la conduite d’un affinement Rietveld sur un composé intermétallique (monophasé) en utilisant le logiciel Fullprof. Il aborde, entre autres, la question des codes d’affinement, de l’ordre d’affinement des paramètres et la détermination des taux d’occupation.
Le fichier de données (.dat) et le fichier initial de description de la structure (.pcr) sont fournis ici.
On suppose un minimum de connaissances de la méthode de Rietveld et du fonctionnement de Fullprof. Si besoin, on peut se reporter à des ressources web sur la méthode de Rietveld, au manuel de Fullprof ainsi qu’à des descriptions et tutoriels disponibles en ligne. On peut voir par exemple le livre « Introduction à la pratique de la diffraction des rayons X par les poudres » de Pierre Gravereau disponible en ligne https://cel.archives-ouvertes.fr/cel-00671294.
Avant de commencer, il est nécessaire d’avoir téléchargé le programme. L’interface de Winplotr permet de travailler en lançant le programme d’affinement puis de visualiser les diagrammes observés et calculés. Il faut aussi se munir d’un bon éditeur de texte. Je conseille Notepad++ qui détecte automatiquement la mise à jour du fichier pcr par Fullprof, et déconseille fortement les outils du type Notepad et WorPad de Windows.
L’exemple concerne le composé LaNi4Pt. Ce composé est un dérivé du composé intermétallique LaNi5, de structure type CaCu5 (groupe d’espace P6/mmm), obtenu par substitution d’une partie du nickel par le platine.
Le composé a été synthétisé par cofusion des éléments purs au four à induction suivi d’un recuit d’homogénéisation. Le composé intermétallique est fragile. On obtient facilement de la poudre par broyage au mortier. C’est cette poudre qui a été mesurée en géométrie Bragg-Brentano avec la radiation du cuivre. Un monochromateur arrière en graphite, capable de supprimer la fluorescence mais non de discriminer les radiations Kα1 et Kα2, a été utilisé. Comme souvent en pareil cas, on ne connaît que la structure du composé parent, ici LaNi5.
Description du fichier .pcr
Fullprof permet de traiter de très nombreux cas dont certains très complexes. L’utilisateur débutant peut être un peu effrayé par la complexité du fichier d’entrée .pcr. Beaucoup des informations contenues dans ce fichier lui seront inutiles. Pour un détail complet, le lecteur est renvoyé vers le manuel de Fullprof (« FullProf_Manual.pdf », disponible dans le dossier « Docs » du répertoire d’installation de Fullprof). Nous proposons ici une description très simplifiée des principaux paramètres.
Le fichier pcr est très pratique car il permet en un seul fichier de donner au programme non seulement toutes les informations structurales mais aussi les instructions sur l’affinement qu’il va conduire.
Tous les paramètres doivent être déclarés. Le programme va lire séquentiellement le fichier .pcr et attendra exactement le nombre de paramètres annoncé par l’utilisateur. Par exemple, si on annonce qu’il y a deux zones exclues en ligne 8 (code Nex à 2), il faut que ces zones apparaissent effectivement (dans notre cas, elles sont en lignes 26 et 27). Si le programme ne trouve pas le nombre exact de lignes qu’il attend, cela conduira à une erreur. Ce type d’erreur est très fréquent chez l’utilisateur débutant. Quand on vient de modifier un fichier qui fonctionnait et qui se met à donner lieu à une erreur, il faut réfléchir aux choses que l’on vient de modifier.
- Nph : il s’agit du nombre de phases (comme pour les zone exclues, le programme attendra le descriptif d’autant de phases annoncées). Ici 1.
- Nba : description du bruit de fond. Ici, 0 veut dire que l’on aura une description polynomiale du bruit de fond.
- Nex : nombre de zones exclues (domaines de 2θ non utilisés pour l’affinement)
- ligne 10 : nom du fichier données xxx.dat
- lambda 1 et lambda 2 sont les longueurs d’onde du doublet Kα1 et Kα2 du cuivre.
- Ratio est le ratio d’intensité entre les deux longueurs d’onde.
- NCY : nombre de cycles d’affinement
- lignes 26-27 : zones exclues. Ici au démarrage, sont définies deux zones exclues de 0° à 0° et de 180° à 180° ce qui évidemment ne sert à rien. Par la suite, on pourra changer les valeurs sans avoir besoin de rajouter des lignes pour couper le début et/ou la fin du diagramme si nécessaire (en mettant une zone exclue de 0° à 15° et une autre de 110° à 180°, par exemple).
- Bkpos : origine du polynôme décrivant le bruit de fond (voir plus loin)
- ligne 30 : nombre de paramètres affinés
Jusqu’à présent les paramètres étaient des paramètres constants. A partir de maintenant, la plupart sont des paramètres variables que le programme va pouvoir ajuster. Chaque paramètre est donc affublé d’un code juste à sa droite pour la ligne 33, juste en dessous pour tous les autres paramètres. Nous reviendrons plus tard sur la syntaxe de ce code. Pour simplifier, au début, disons que si la valeur de ce code est nulle, le paramètre n’est pas ajusté. Si on souhaite l’ajuster, il faut donner au paramètre un numéro, suivi d’un 1 pour que Fullprof comprenne qu’il faut affiner ce paramètre. Les numéros des paramètres doivent être donnés dans l’ordre. Au cours d’un affinement, il faut tenir à jour le nombre de paramètres que l’on affine. Il faut faire attention de ne pas donner le même numéro à deux paramètres différents et à ne pas laisser de trou entre les numéros. Si le Number of refined parameters de la ligne 30 est égal à 4, le programme attendra 4 paramètres numérotés de 1 à 4, suivis d’un 1. Cette contrainte ne pose en général pas trop de soucis car l’idée est d’ajouter les paramètres au fur et à mesure de manière à avoir une optimisation tournant à la fin avec tous les paramètres en même temps. D’autre part, il est bon de vérifier l’évolution des variables en cours d’affinement pour s’assurer que les valeurs obtenues ont un sens physique (Fullprof n’a pas forcément de garde-fous sur certaines variables qui peuvent prendre des valeurs irréalistes). C’est à l’utilisateur d’être critique sur les valeurs obtenues en cours d’affinement.
- Ligne 35 : paramètres décrivant le bruit de fond. Si on a choisi la description polynomiale du bruit de fond, on trouve sur cette ligne les paramètres b0 à b6 avec lesquels sera décrit le bruit de fond selon l’équation :
Jusqu’ici, les paramètres étaient des paramètres généraux. A partir de maintenant, on a une description de la structure de chacune des phases, phase par phase.
Dans notre exemple, nous n’avons qu’une seule phase de structure CaCu5. Cette structure a été reprise directement de la littérature pour le composé LaNi5. Les sources peuvent être nombreuses : publications ou bases de données cristallographiques (Crystallography Open Database http://crystallography.net/cod/browse.html, Pearson’s Handbook of Crystallographic Data for Intermetallic Phases, Pearson Crystal Data http://www.crystalimpact.com/pcd/, Inorganic Crystal Structrue Database https://icsd.products.fiz-karlsruhe.de). Cette description ne correspond pas exactement à notre composé mais nous verrons plus loin si cela peut constituer une base suffisamment solide pour démarrer l’affinement.
- Nat: nombre d’atomes. C’est en fait le nombre de lignes de description atomique. Il correspond au nombre de sites occupés. Mais si plusieurs atomes de nature différente occupent le même site, les lignes devront être dédoublées pour chaque type d’atome. Les choses vont s’éclaircir avec l’exemple.
- Ligne 51 : groupe d’espace, ici P6/mmm (groupe n°191). Une table de correspondance est disponible à cette adresse.
- Lignes 53, 55 et 57 : descriptif de chaque atome. On doit donner les coordonnées atomiques, le facteur de déplacement et l’occupation (Occ). Dans notre cas, dans la structure CaCu5, il y a trois sites occupés. Le La occupe la position 1a (0,0,0), le Ni les positions 2c (1/3,2/3,0 ; 2/3,1/3,0) et 3g (1/2,0,1/2 ; 0,1/2,1/2 ; 1/2,1/2,1/2). Evidemment, grâce au groupe d’espace, le programme génère automatiquement les atomes équivalents par symétrie (par exemple l’atome de Ni en 2/3, 1/3, 0 à partir de la donnée de celui en 1/3,2/3,0). En revanche, les occupations sont à donner en nombre d’atomes vrais. C’est-à-dire qu’il faut mettre 2 pour la position 2c et 3 pour la position 3g. La lecture du pcr donne donc le contenu de la maille (ici 1 La et 2+3 Ni). Cela diffère d’autres programmes pour lequel il faut donner la fraction de site (ici, ce serait 1 pour chaque position).
- Scale : le facteur d’échelle. Ce facteur permet de mettre à l’échelle les intensités calculées par le programme avec la mesure réelle en nombre de coups qui dépend de tout un nombre de paramètres inconnus du programme (quantité de poudre, temps d’acquisition, intensité incidente etc…).
La fonction que nous utilisons pour décrire la forme des raies est la fonction #5 (Pseudo-Voigt, Npr = 5 ; attention présent plusieurs fois, dans les codes d’en-tête et dans la description de la phase). Elle se décrit mathématiquement comme une combinaison linéaire d’une fonction gaussienne et d’une fonction lorentzienne de même largeur H. La fonction ne sépare pas les contributions instrumentales et échantillon à la largeur de raies. Le coefficient de mélange est η qui doit normalement varier entre 0 (pure gaussienne) et 1 (pure lorentzienne).
La largeur H est, elle, décrite en utilisant la fonction de Caglioti :
Le paramètre shape1 correspond à η. Les paramètres U, V et W sont clairement indiqués dans le pcr.
- Ligne 66 : les paramètres de maille et les angles (noter l’angle de 120° pour la structure hexagonale).
Examen de la correspondance entre le modèle structural entré et la mesure
On fait tourner le programme avec le fichier .pcr et le fichier de données .dat. Aucun paramètre n’est affiné à ce stade (nombre de paramètres affinables = 0). On effectue juste une comparaison entre ce que donne le modèle structural et les données. Cette première étape est essentielle. Un examen approfondi du résultat doit guider l’utilisateur sur la conduite à tenir pour presque tout l’affinement.

Figure 1
Voilà ce que l’on obtient après un zoom (Fig. 1). Que voit-on ?
Les données sont sous la forme de points rouges. La courbe calculée est sous la forme d’un courbe noire, la différence entre les deux courbes sous la forme d’une courbe bleue. Les petits bâtons bleus représentent les positions des réflexions générées par le groupe d’espace et dont la position est fixée par les paramètres de maille. Toutes les positions sont dédoublées, correspondant au doublet Kα1 et Kα2.
A ce stade, après différents zooms, nous pouvons faire les conclusions suivantes :
- la structure que nous avons supposée semble être la bonne. Il y a une assez bonne ressemblance en termes de présence des pics et d’intensité relative des pics entre les courbes calculée et observée
- on ne met pas en évidence de pics supplémentaires indiquant qu’il pourrait y avoir une autre phase
- en revanche les pics sont très décalés en position (vers les plus grands angles) et les intensités sont globalement beaucoup plus importantes pour la courbe calculée
Avant de se lancer dans le processus d’affinement, il est bon de garder en tête quelques bonnes pratiques. Il est notamment conseillé de faire des sauvegardes fréquentes du fichier .pcr car avec l’option que nous avons choisie (Pcr = 1), celui-ci est mis à jour par le programme automatiquement à chaque cycle (code NCY). Une sécurité existe toutefois. Le programme compare, à la fin d’un cycle d’affinement, la valeur du χ2 avec celle obtenue au cycle précédent et qui figure en ligne 2. Une valeur supérieure indique une divergence et dans ce cas le pcr n’est pas mis à jour. Ce sera clairement indiqué dans la fenêtre du déroulement de l’affinement si l’on est confronté à ce problème. D’ailleurs quand on repart d’un ancien fichier pcr, pour affiner un nouveau diagramme, il ne faut pas oublier d’augmenter énormément cette valeur de χ2 sinon le programme ne commencera jamais à affiner les variables (ainsi que de remettre à 0 tous les codes d’affinement).
Premiers pas dans l’affinement
A ce stade, après analyse de la différence entre les courbes observées et calculées, il faut se demander ce que l’on peut faire pour améliorer l’accord. Les questions sont un peu toujours les mêmes :
- d’où viennent les désaccords ?
- d’où vient le modèle calculé que l’on utilise et quelles peuvent être les différences entre ce modèle et celui qui représenterait le mieux notre échantillon ?
- quels paramètres doivent être changés pour améliorer l’accord ?
- quel est le paramètre qui va permettre la plus grande amélioration de l’accord à ce stade ?
- doit-on laisser le programme ajuster ce paramètre ? Ou peut-on/doit-on le faire manuellement ?
Dans notre cas, il est évident que l’accord est très mauvais. Probablement trop mauvais pour laisser le programme travailler seul à ce stade. N’oublions pas que dans une méthode de moindres carrés non linéaire, il faut partir de paramètres proches des paramètres finaux. La méthode de Rietveld est une méthode d’affinement ou d’ajustement, pas vraiment une méthode de détermination des paramètres structuraux. Les paramètres structuraux qui peuvent être déterminés sans connaissance préalable de leur valeur sont des paramètres qui n’influent que modérément sur la courbe calculée.
Dans notre cas, les paramètres de maille sont tellement éloignés des paramètres réels qu’il est impossible de les ajuster. Si notre œil est capable de voir aisément qu’il suffit de décaler tous les pics vers les bas angles pour superposer les courbes observés et calculés, le programme, lui, en est incapable. Il y a trop peu de recouvrement entre les pics observés et calculés pour que le programme trouve qu’en augmentant les paramètres de maille, il va améliorer l’ajustement.
Ce serait aussi le cas des positions atomiques si elles étaient inconnues. La détermination structurale, si le type structural est inconnu, doit être faite avec d’autres outils. En revanche, nous verrons plus tard que l’on peut ajuster les taux d’occupation car leur influence sur le calcul est plus modérée.
Il faut aussi réfléchir à notre modèle et à notre échantillon. Faute de données dans la littérature sur notre composé (LaNi4Pt), nous avons choisi d’introduire les données structurales d’un composé proche : LaNi5. Le platine étant nettement plus gros que le nickel qu’il remplace, il n’est pas vraiment étonnant que les paramètres de maille soient plus grands (les pics décalés vers les bas angles).
On ne peut pas laisser le programme les ajuster. Il faut modifier ces paramètres de maille soi-même. A ce stade, on a plusieurs options plus ou moins intelligentes, efficaces ou rapides à mettre en œuvre : utiliser un programme extérieur de détermination des paramètres de maille en fonction de la position des pics, utiliser un logiciel d’indexation par comparaison avec des bases de données (type EVA ou HighScore utilisant le Powder Diffraction File), bouger soi-même les paramètres de maille à la main et voir comment les pics se déplacent, déterminer soi-même une valeur approchée de a et c à partir des positions des deux pics (110) et (002) qui ne dépendent, chacune, que d’un seul paramètre…
Personnellement, étant plutôt faignant, je préfère la méthode pas très intelligente mais rapide consistant à regarder les distances interréticulaires d mesurées et calculées données par Winplotr sur une position. Si on prend la raie (111) elle est calculée pour d=2.123 Å alors que la raie observée est à 2.150 Å. On en déduit que grossièrement si on multiplie a et c par 2.150/2.123=1.013, on devrait obtenir quelque chose de plus satisfaisant. On met donc a=b=5.020×1.013=5.084 Å et c=4.032 Å dans le fichier .pcr et on refait tourner.

Figure 2
Le résultat n’est pas parfait (Fig. 2). Il y a encore des décalages probablement dû au fait que a et c n’ont pas augmenté de manière isotrope lors de la substitution (on apprend déjà des choses sur notre échantillon !). Mais au moins, maintenant, il y a un bon recouvrement des pics, probablement suffisant pour laisser le programme ajuster les paramètres.
A ce stade, il convient de se poser les mêmes questions que précédemment. Quel est le paramètre qui va le plus améliorer notre affinement ? Il était évident au stade précédent qu’il fallait modifier les paramètres de maille. Il est tout aussi évident maintenant que ce n’est plus la priorité. Le plus grand désaccord maintenant provient du fait que les intensités calculées sont toutes beaucoup plus grandes que les intensités observées et cela de manière systématique. Il faut effectuer une mise à l’échelle entre les diagrammes. C’est le facteur d’échelle (scale) que l’on doit maintenant ajuster. Peut-on laisser le programme le faire, même si la valeur finale est manifestement assez loin de la valeur actuelle ? Cette fois, la réponse est oui. Si on ajuste que ce seul paramètre le programme ne peut faire autre chose que de le diminuer pour améliorer l’accord. Ce n’était pas le cas tout à l’heure pour les paramètres de maille. On peut aussi travailler par ajustement manuel en essayant par exemple de le régler manuellement, mais c’est une perte de temps dans notre cas. A noter que si, tout à l’heure, on avait commencé par affiner le facteur d’échelle alors que les paramètres de maille étaient mal ajustés, le programme aurait beaucoup trop diminué ce paramètre car il n’y avait pas d’intensité sur les positions des pics calculés. Maintenant que les paramètres de maille sont à peu près réglés, on peut affiner ce paramètre. Alors qu’à l’inverse, maintenant, affiner les paramètres de maille ne donnerait rien avec un facteur d’échelle aussi mal ajusté. Tout cela, pour montrer les corrélations entre paramètres.
On met donc 1 paramètre affiné et on introduit le code 11.0000 sous le code Scale.

Figure 3
Affinement du bruit de fond
A ce stade (Fig. 3), on constate que la chose la plus mal décrite maintenant est le bruit de fond. On rappelle ici que le facteur d’accord χ2, qui est la valeur guidant le programme dans sa minimisation, est calculé sur l’ensemble du diagramme (pas seulement sur les pics) en faisant intervenir la somme des carrés des différences entre les intensités calculées et observées pour chacun des points du diagramme. Il est donc extrêmement important de bien ajuster le bruit de fond dès le début de l’affinement.
Nous avons choisi une description polynomiale. Si on choisit une description de bruit de fond pointé, il est facile d’utiliser Winplotr pour sélectionner des points dans le bruit de fond (choisir des points loin des pics et décrivant une courbe bien régulière). On les sauve et on les copie-colle dans le fichier .pcr en n’oubliant pas de mettre le bon nombre de ligne pour le paramètre Nba.
Dans notre cas, nous avons un polynôme. On peut laisser le programme se débrouiller mais il est aussi très simple de donner une valeur pour le paramètre b0. En zoomant dans Winplotr, on voit que le bruit de fond est très constant autour d’une valeur proche de 80. On peut introduire cette valeur dans le .pcr.
Affinement des paramètres de maille
A ce stade, en faisant différent zooms, on voit qu’il y a des problèmes sur la forme des pics et sur la position des pics, surtout aux grands angles. On doit comprendre qu’il n’est pas possible ni raisonnable d’affiner la forme des pics tant que les positions entre pics calculés et observés ne correspondent pas. En revanche, la mauvaise description des largeurs ne gêne pas pour affiner les positions. C’est ce genre de corrélations entre les paramètres que doit peu à peu appréhender le nouvel utilisateur.
On affine a et c en mettant 3 paramètres affinés et les codes 21.0000 sous a et b et 31.0000 sous c. Le fait d’utiliser le même code pour a et b est nécessaire pour garder les deux égaux. La même variation va être appliquée à chaque cycle d’affinement pour les deux paramètres. Une petite complexité pour Fullprof vient du fait qu’il faut mettre aussi le code 21.0000 pour le paramètre gamma. Cela est nécessaire uniquement pour les structures hexagonales (voir le manuel de Fullprof pour une explication pas très facile à comprendre, mieux vaut accepter de faire çà sans trop chercher à comprendre). Il est possible que 10 cycles d’affinement ne suffisent pas à bien caler les positions des pics et à faire converger le programme. On peut le relancer plusieurs fois si besoin ou augmenter le nombre de cycles d’affinement (NCY). Cela revient au même.

Figure 4
Même après convergence (Fig. 4), on observe toujours des décalages. Il est utile d’affiner conjointement tous les paramètres ayant une influence sur les positions des pics. On peut donc rajouter le décalage instrumental (zero). On lui met le code 41.000 (à sa droite) et on n’oublie pas de changer le Number of refined parameters à 4.
On a maintenant un bon accord de position même à grand angle (Fig. 5) :
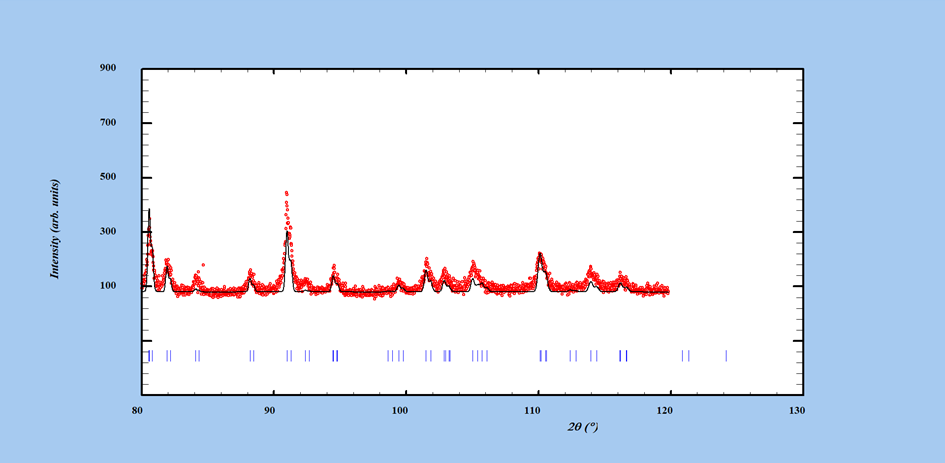
Figure 5
En effet la valeur trouvée (zero = 0.178) est très importante et provoquait l’impossibilité d’ajuster les positions avec seulement a et c.
Affinement des profils de raies
Il est temps maintenant de s’attaquer aux profils de raies (Fig. 6) :

Figure 6
De nouveau, réfléchissons au paramètre qui peut améliorer l’ajustement. Dans le profil de raies, il y a deux composantes : la largeur à mi-hauteur et la forme elle-même avec le pourcentage gaussien/lorentzien. On voit que la largeur à mi-hauteur n’est pas forcément si mal ajustée, en revanche l’examen des pieds de pics tombant beaucoup trop vite indique que les raies calculées sont beaucoup trop gaussiennes (de fait le paramètre η (shape1) est fixé à 0.1). Affinons-le (paramètre 5 pour le shape, Number of refined parameters = 5).
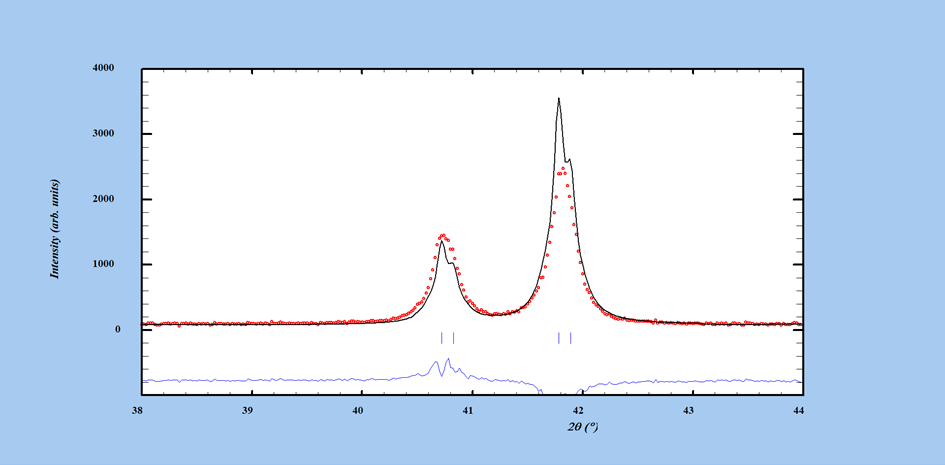
Figure 7
La description est bien meilleure (Fig. 7) mais le paramètre a convergé vers des valeurs irréalistes car supérieures à 1. Ce peut être sous l’influence des largeurs qui sont mal ajustées. On peut alors soit essayer de rajouter les largeurs de raies, soit fixer η à un, affiner les largeurs et remettre η en plus des largeurs quand elles seront ajustées. On prend cette seconde option même si, en principe, on ne retire pas de l’affinement un paramètre déjà affiné. On met 5 pour W et 6 pour U en fixant shape1 à 1. On aura alors une description simplifiée de la fonction de Caglioti avec un terme V nul.
C’est le moment d’évoquer les problèmes que l’on peut avoir avec l’affinement des largeurs de raies qui est quelquefois délicat. On peut avoir une divergence du programme ou un message d’erreur indiquant que les largeurs au carré sont négatives. Ces problèmes viennent du fait que :
- ce ne sont pas directement les largeurs qui sont ajustées mais le carré des largeurs à travers la fonction de Caglioti présentée plus haut. Une combinaison accidentelle des valeurs de U, V et W produisant pour certaines valeurs de θ ou de tg θ des valeurs négatives pose effectivement des problèmes numériques
- la fonction de Caglioti avec trois paramètres est un peu complexe et peut causer des problèmes notamment si il y a peu de réflexions et si le domaine angulaire des données est relativement réduit
- l’affinement est très sensible au choix des valeurs initiales
Comme (presque) toujours, un peu de bon sens permet de résoudre les problèmes. On peut lister quelques conseils :
- ne pas entreprendre l’affinement des largeurs avant d’avoir bien ajusté un certain nombre de paramètres et d’avoir déjà une description satisfaisante du diagramme. Par exemple, il faut au minimum que les raies soient au bon endroit.
- ajuster soi-même les valeurs initiales de U, V et W si l’accord entre les largeurs calculées et observées n’est vraiment pas bon. On peut au besoin partir avec U=0, V=0 et W fixé à la racine carrée de la largeur à mi-hauteur à bas angle que l’on peut facilement mesurer soi-même. Pour des cas très difficiles, on peut aussi mesurer soi-même et faire une liste des largeurs au carré en fonction de la tangente de l’angle, voir la tendance et faire un ajustement soi-même dans Excel par exemple pour trouver des bonnes valeurs de départ de U et W, éventuellement de V.
- commencer par affiner W seul puis U pour tenir compte de l’élargissement. Souvent ces deux paramètres sont suffisants. Si c’est nécessaire, si les données sont de bonnes qualités et/ou s’étendent très loin en angle (120° à 140° 2θ), on peut rajouter V. Si un problème se pose, ne pas hésiter à remettre V à zéro et à le fixer.
- en dernière extrémité, par exemple si le polynôme de Caglioti s’extrapole mal aux bas angles donnant des valeurs négatives pour les toutes premières réflexions, il est toujours possible de fixer définitivement des paramètres comme W ou U. On peut vérifier, bien sûr, que l’accord de la description reste correct dans l’ensemble du diagramme.
Dans notre exemple, l’affinement se passe plutôt bien. On remet η avec le paramètre 7. On obtient maintenant 1.04 pour η ce qui est acceptable. Cela montre la corrélation qui existait entre les largeurs et ce paramètre. On essaie d’affiner le V avec le paramètre 8. L’affinement converge sans problème (Fig. 8). Affiner brutalement U, V et W à l’une des étapes précédentes aurait probablement conduit à une divergence.

Figure 8
Fin de l’affinement, ajustement des intensités
Observons à nouveau nos diagrammes observés et calculés. Nous n’avons pas insisté sur ce point mais il est souvent très utile d’observer aussi la courbe différence. Un décalage de position (Fig. 4) ou des largeurs mal ajustées (Fig. 6) y laissent des formes caractéristiques. Ici, on peut constater facilement que le gros défaut est maintenant sur les intensités. Certains pics calculés sont beaucoup trop intenses, d’autres beaucoup trop faibles.
Examinons les paramètres pouvant influencer le calcul des intensités :
- les facteurs de déplacement B
- les positions atomiques
- la nature des atomes et les taux d’occupation
Il n’existe pas de corrélation de nos différences d’intensité en fonction de l’angle (deux pics proches peuvent avoir des différences d’intensité contraires). Si, au contraire, on avait eu tous les pics aux bas angles trop faibles et tous les pics aux grands angles trop intenses, cela aurait été caractéristique d’un problème sur les facteurs de déplacement (B) car, d’après la formule du facteur de structure, de grandes valeurs de B diminuent les intensités à grand θ. Ce n’est pas notre cas, les facteurs B à ce stade ne doivent pas jouer un rôle prépondérant.
Dans la structure, il n’y a pas de positions atomiques variables (que l’on puisse affiner). Elles sont toutes fixées par la symétrie.
Le problème vient donc probablement de la nature des atomes sur les sites ou de leur taux d’occupation. Là encore, il est aisé de se rendre compte que jusqu’à maintenant nous avons affiné la structure du composé LaNi4Pt avec la structure de LaNi5. Notre modèle ne prend pas en compte le platine en substitution au nickel. On peut imaginer pourtant que l’effet du platine (ZPt=78, ZNi=28) va être très important sur le facteur de structure et les intensités à cause de la différence importante du nombre d’électrons. Il faut donc nécessairement en tenir compte. Il y a deux sites pour le nickel. Nous n’avons aucune information sur le fait que le platine va se localiser préférentiellement sur l’un ou l’autre des sites. Le meilleur moyen de le savoir est justement d’utiliser la méthode de Rietveld !
On pourrait faire des essais en plaçant alternativement le Pt sur un site, puis sur l’autre puis de manière statistique mais autant laisser le programme travailler.
Pour cela nous allons répartir le platine sur les sites de la façon suivante :
La LA 0.00000 0.00000 0.00000 1.50000 1.00000 0 0 0 0 0.00 0.00 0.00 0.00 0.00 Ni1 NI 0.33333 0.66666 0.00000 1.50000 1.50000 0 0 0 0 0.00 0.00 0.00 0.00 91.00 Ni1 PT 0.33333 0.66666 0.00000 1.50000 0.50000 0 0 0 0 0.00 0.00 0.00 0.00 -91.00 Ni2 NI 0.50000 0.00000 0.50000 1.50000 2.50000 0 0 0 0 0.00 0.00 0.00 0.00 -91.00 Ni2 PT 0.50000 0.00000 0.50000 1.50000 0.50000 0 0 0 0 0.00 0.00 0.00 0.00 91.00
Nous avons rajouté deux lignes aux mêmes positions que les deux atomes de nickel en changeant la nature de l’atome Ni pour Pt. Nous avons maintenant 5 lignes de descriptions des atomes et il faut vite changer le paramètre Nat de 3 à 5 avant d’oublier de le faire.
Nous arrangeons ensuite les paramètres occ de manière à avoir la bonne composition. Nous avons un platine par formule. Il y a une formule par maille. Nous pouvons, arbitrairement, et comme valeur de départ, placer 0.5 Pt sur le site 2c et 0.5 Pt sur le site 3g. L’important est d’avoir le bon nombre total. La distribution, elle, va être ajustée. Ce nombre total correspond à la stœchiométrie nominale et à l’analyse qui a pu être faite par microsonde. Nous avons donc confiance en la composition et nous ne cherchons pas à l’affiner (ce qui serait tout à fait possible d’ailleurs). Le platine est en substitution, il faut donc retirer le nombre de nickel correspondant sur chaque site. Au final, nous avons 1.5 Ni sur 2c et 2.5 Ni sur 3g.
Maintenant, il nous faut revenir sur les codes d’affinement. Nous avons dit plus haut, pour simplifier, que le code 91.0000 se composait du numéro du paramètre (ici 9) suivi de 1 si on souhaitait affiner ce paramètre. En fait, c’est un peu plus subtil. Le 1 est à comprendre comme un facteur multiplicatif qui va être appliqué au déplacement (shift) calculé par le programme sur le paramètre 9. La nouvelle valeur pour les variables associées au paramètre 9 seront l’ancienne valeur plus le déplacement calculé multiplié par le facteur multiplicatif.
Prenons un exemple pour clarifier. Dans beaucoup de structures hexagonales, il y a des positions x,2x,z. Quand on affinera les positions dans une telle structure, il faudra veiller à ce que la valeur de y soit toujours égale à deux fois la valeur de x. Le seul moyen d’y arriver est, d’une part, d’avoir initialement la condition respectée et, d’autre part, de faire en sorte que les déplacements sur les variables soient répercutés avec un facteur 2 sur la valeur y. Par exemple :
Ni1 NI 0.1000 0.20000 0.00000 1.50000 1.50000 0 0 0 0 91.00 92.00 0.00 0.00 0.00
C’est ce qui se passera si on utilise la variable 9 avec un coefficient multiplicateur de 1 pour x et de 2 pour y. Lors d’un cycle d’affinement, un shift calculé de 0.005 sur la variable 9 sera appliqué à x et multiplié par 2 avant d’être appliqué à y. Les nouvelles valeurs seront 0.105 et 0.210. Le rapport deux entre x et y est ainsi conservé.
Si on revient à notre exemple, nous souhaitons affiner un atome en substitution, donc si un ajout de platine est fait par le programme, il faut qu’une quantité équivalente de nickel soit retirée. En plus, comme nous souhaitons conserver la composition, il faut aussi retirer le platine de l’autre site et le compenser aussi par le nickel. C’est ce qui explique les codes 91 et -91 tels que montrés plus haut.
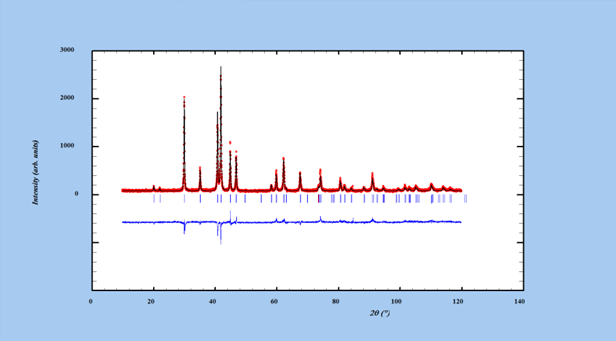
Figure 9
Une amélioration significative est notée (Fig. 9). Cette fois la courbe différence fait apparaître des disparités notables entre les bas angles et les grands angles que l’on peut associer à mauvais ajustement des B.
On peut dans un premier temps ajuster le même B pour tous les atomes (paramètre 10).

Figure 10
L’ajustement est maintenant relativement satisfaisant (Fig. 10). On peut maintenant examiner les paramètres. Le platine est entièrement localisé sur le site 3g (ce qui est en accord avec sa taille plus grosse que le Ni). Le paramètre η à 1.10 est un peu grand (raies super-lorentziennes). Cela arrive fréquemment avec les composés intermétalliques. Cela est due à la présence au sein de la poudre de plusieurs niveaux de cristallinités différents. On peut éventuellement le fixer à 1. Il arrive fréquemment que les paramètres B soient affinés négatifs pour des composés très fortement absorbants des rayons X (c’est le cas des métaux). Il ne faut pas trop s’en inquiéter car ce paramètre recouvre à la fois les vrais B mais aussi la composante de micro-absorption (facteurs de Brindley) qu’il est impossible de dissocier. Il est juste de faire l’affinement comme çà, en revanche, il faut être conscient que les valeurs de B sont fausses.
Conclusion
Pour finir, quelques conseils :
- la comparaison initiale, sans ajustement, est vraiment très importante : la conduite de l’affinement en dépend
- elle dépend donc évidemment du choix qui a été fait pour le modèle structural utilisé. Plus il est proche du modèle réel de notre échantillon, plus l’affinement sera facile. En général, on fait toujours un affinement en utilisant le .pcr final d’un affinement précédent et que l’on modifie. Il fait faire très attention à ce stade de bien choisir ce .pcr de manière à ce que les paramètres décrivent le mieux possible notre situation présente et à l’adapter au nouveau cas de manière intelligente.
- l’ordre dans lequel on affinera les paramètres dépend évidemment de la qualité de l’accord initial et de la proximité entre le modèle structural choisi au départ et le modèle final. A titre d’exemple, si on choisit un prototype dont les paramètres de maille correspondent exactement aux paramètres de notre échantillon, il ne sera évidemment pas nécessaire de les ajuster en premier.
- vérifier à chaque étape le sens physique des variables : η, B, taux d’occupation, coordonnées atomiques
- quand le programme fait des choses qui ne semblent pas normales, se souvenir que le programme cherche à minimiser globalement les écarts observés-calculés sur l’ensemble du diagramme. Il peut ainsi dégrader l’affinement de la phase d’intérêt si c’est au profit d’un meilleur ajustement de l’ensemble du diagramme. C’est la raison pour laquelle, pour obtenir un bon ajustement de la phase d’intérêt, il faut aussi bien ajuster tout le reste (bruit de fond, profils, introduction des phases supplémentaires…)
- garder à l’esprit les corrélations entre les paramètres et affiner les paramètres corrélés plus ou moins en même temps.
- les paramètres de maille avec le décalage de zéro qui influent sur la position des raies
- la composante gaussienne/lorentzienne avec les largeurs de raies qui influent sur les forme des pics
- les positions atomiques, taux d’occupation et facteurs de déplacement qui influent sur les intensités des pics
Quelques erreurs fréquentes :
- hole in the normal matrix : il manque un numéro dans les paramètres affinés. Vérifier tous les paramètres et les renommer pour éviter le trou dans la liste.
- integer in a numeral field : l’erreur indiquée par le programme n’est pas forcément là où l’erreur a été commise. Le fichier .pcr est lu séquentiellement. Le programme attend exactement le nombre de ligne ou de paramètres déclaré. Repenser à la dernière modification faite. Cela arrive souvent quand on a ajouté ou supprimé des phases, des atomes, des points de bruits de fond ou des zones exclues
- vérifier souvent les numéros des paramètres affinés et vérifier que l’on n’a pas attribué le même numéro à des paramètres différents.