
Olivier RAPAUD – IRCER, UMR CNRS 7315, Université de Limoges
Jean-Marc JOUBERT – ICMPE, UMR CNRS 7182, Université Paris-Est Créteil
Préambule
Le terme CALPHAD (CALculation of PHAse Diagrams) reviendra souvent par la suite. Ce n’est pas un logiciel, ni une base de données. Il s’agit d’une méthode de modélisation des fonctions thermodynamiques (principalement de l’enthalpie libre, toutes les autres fonctions thermodynamiques dérivant de G) caractérisant les corps purs, les composés, et les solutions (solides, liquides et gazeuses).
Il faut bien comprendre que la méthode CALPHAD peut s’utiliser de deux manières différentes suivant les utilisateurs.
Certains utilisent la méthode pour obtenir la description thermodynamique d’un système. Les données d’entrée sont les données expérimentales ou calculées sur ce système. L’opération consistant à mettre en équation le système, c’est-à-dire déterminer les paramètres décrivant les énergies de Gibbs des différentes phases s’appelle optimisation thermodynamique. Ces descriptions sont alors stockées dans des bases de données où elles peuvent être combinées avec des descriptions d’autres systèmes.
D’autres utilisateurs de la méthode font l’opération inverse, c’est-à-dire qu’ils utilisent des bases de données déjà constituées pour faire des calculs thermodynamiques ou des calculs d’équilibre dans des systèmes qui peuvent être simples ou très complexes. Cette restitution est possible et juste, pour les systèmes simples, si les systèmes ont été bien décrits dans la première étape, et donc s’ils bénéficiaient d’un grand nombre de données expérimentales cohérentes. Si c’est le cas, le calcul permet de restituer non seulement les données qui étaient connues mais aussi un grand nombre de grandeurs qui ne l’étaient pas forcément. A titre d’exemple, un système binaire a pu être modélisé en utilisant des enthalpies de formation connues pour seulement certaines phases, alors que le calcul pourra donner les enthalpies de formation de tous les composés et même les enthalpies de mélange et les activités dans toutes les solutions. Pour les systèmes complexes qui n’ont pas forcément été modélisés en tant que tel, mais dont certains sous-systèmes l’ont été, le calcul se base sur les combinaisons des énergies de Gibbs des différentes phases permettant de faire des extrapolations dans les systèmes multi-constitués. Ces calculs prédictifs sont très souvent extrêmement justes dans des systèmes d’alliages industriels pouvant aller jusqu’à une dizaine d’éléments même si seulement les sous-systèmes binaires et quelques ternaires d’intérêt ont été optimisés.
Dans cette page, nous aborderons de manière plus précise la première partie, c’est-à-dire l’étape de modélisation ou d’optimisation proprement dite.
1. Introduction
La volonté de vouloir décrire les corps purs, les composés et les solutions n’est pas nouvelle [1]. Elle est à la base de la méthode CALPHAD [2,3]. Cette méthode repose sur la description des constituants par complexité croissante (en partant des corps purs dans tous leurs états d’agrégation possibles) sur la base de l’expression polynomiale de l’enthalpie libre G du corps pur ou de la phase considérée. Dans cette expression, des variables seront ajustées à partir de données expérimentales par une méthode de moindres carrés.
2. Définition d’un état de référence : l’état SER
L’enthalpie n’étant pas une grandeur absolue mais relative, il faut donc définir un état de référence. Les premières générations de bases de données (1 et 2) définissent l’état de référence à 298,15 K à la pression standard (1 bar, soit 105 Pa) pour plusieurs raisons: d’abord pratiques, puisqu’expérimentalement la quasi-totalité des calculs se faisaient à bien plus hautes températures, mais aussi pragmatiques car les puissances de calcul des ordinateurs de l’époque n’autorisaient pas vraiment des calculs dès 0 K (à part peut-être pour l’énergie de formation). Cet état standard de référence est appelé SER pour « Standard Element Reference », c’est-à-dire la phase stable (structure cristallographique) à 105 Pa et 298,15 K.
Le problème majeur de cet état de référence à 298,15 K est qu’il ne permet pas de rejoindre la physique à basse température. En effet, avec les bases de données de génération 1 ou 2, l’extrapolation de la capacité calorifique, par exemple, à 0 K donne des valeurs non nulles (et il en va donc de même pour l’entropie), ce qui viole la troisième loi de la thermodynamique. Actuellement, un consortium existe visant à l’utilisation d’un formalisme plus adapté de description de l’enthalpie libre des corps purs en fonction de T dès 0 K. On l’appellera « de troisième génération ». Nous l’évoquerons un peu plus loin.
3. Description des corps purs
Si nous revenons à la description polynomiale de l’énergie de Gibbs en fonction de la température G(T) du corps pur m en utilisant un formalisme 2ème génération, à l’état d’agrégation stable dans les conditions standard de référence, il vient:
Mais pour aller plus loin que cette simple considération « pratique », notamment pour les corps purs à 298,15 K dans leur structure cristalline stable pour lesquels l’enthalpie est définie comme nulle, il est également nécessaire d’introduire une autre notion importante: la stabilité de réseau (ou lattice stability en anglais). Pour un même corps pur, toutes les variétés cristallines (allotropes) doivent être décrites par rapport à cet état de référence. Ceci a été fait dès 1991 par A.T. Dinsdale dans son article « SGTE Data for pure elements » [4] où il décrit chaque constituant dans la phase φ par rapport à son état de référence par l’expression . Nous reviendrons sur cette notion de stabilité de réseau par l’exemple un peu plus loin lorsque nous traiterons des solutions.
Pour comprendre d’où provient le polynôme utilisé, partons d’une donnée expérimentale très importante en modélisation thermodynamique: la capacité calorifique Cp. En partant de la relation de Maier-Kelly pour l’expression du Cp:
Par intégration de Cp en fonction de T, il vient:
Et par intégration de Cp/T, on obtient:
Ainsi, l’expression de G devient:
Qu’on simplifie bien souvent par:
C’est cette différence que l’on retrouve sous la dénomination GHSER dans les bases de données. On remarque que le terme A est purement enthalpique (constante d’intégration a du Cp par rapport à T), alors que B contient une contribution entropique b et le terme indépendant de T du Cp, c. Les autres variables (C, D, E, et F) correspondent à une contribution vibrationnelle du Cp.
Ce qu’il faut retenir c’est que les descriptions polynomiales G(T) sont compilées pour tous les corps purs dans une base de données librement téléchargeable et que toute la communauté utilise: il s’agit de la base des corps purs, nommée PURE, produite et maintenue par le SGTE (Scientific Group Thermodata Europe). Cette base contient également les descriptions des corps purs dans des états d’agrégation différents (stables, métastables) de celui correspondant à l’état stable dans les conditions standard de référence. Ainsi, cette base des corps purs contient également toutes les énergies de stabilité de réseau par rapport à l’état standard de référence.
4. Description des composés définis
Après avoir évoqué les corps purs, nous pouvons maintenant traiter les composés définis.
L’enthalpie libre des composés définis est uniquement dépendante de la température. En effet, la composition est connue et fixe. Si l’évolution de la capacité calorifique en fonction de la température du composé d’intérêt est connue, ainsi que son enthalpie de formation, on peut la décrire de la même manière qu’un corps pur.
Si malheureusement l’évolution de sa capacité calorifique n’est pas connue, il est possible en première approximation d’utiliser une loi de mélange, connue sous le nom de règle de Neumann-Kopp, appliquée au mélange des corps purs constitutifs du composé défini. La capacité calorifique d’un composé stœchiométrique est dans ce cas la somme pondérée des capacités calorifiques des éléments purs. Il vient alors: . Son utilisation dans la description polynomiale de G(T) s’écrit alors:
Dans ce cas, et dans cette approximation de Neumann-Kopp, l’enthalpie de formation du composé à partir des éléments purs dans leur état SER est a et son entropie de formation -b. Ces deux grandeurs sont alors indépendantes de la température.
5. Description des solutions solides désordonnées : les solutions de substitution
Nous allons commencer à traiter des solutions en partant peut-être de la plus simple à appréhender. Dans le cas d’une solution de substitution, des atomes de nature différente se substituent sur les positions d’un même réseau cristallin.
Si on revient à l’expression polynomiale de l’énergie de Gibbs de la phase φ correspondant à une solution de substitution, celle-ci se décompose de la manière suivante:
On voit ici apparaître trois contributions (il en existe d’autres, comme une contribution magnétique, mais nous n’en parlerons pas ici).
La première, , est l’énergie de Gibbs dite de référence, qui est une combinaison linéaire des enthalpies libres des constituants purs dans la phase considérée pondérées par la fraction molaire de chaque constituant. On remarque ici qu’il est nécessaire de connaitre l’enthalpie libre des constituants (corps purs) dans un état φ qui peut ne pas correspondre à l’état stable à l’état standard. On retrouve de nouveau cette notion d’énergie de stabilité de réseau.
On peut noter la similarité de cette équation avec celle donnée précédemment pour un composé défini. Cependant, il y a deux différences majeures: ici la composition est variable (la solution est définie pour toute valeur de x entre les deux constituants A et B) et le G de référence n’est plus nécessairement le GSER mais, pour les deux éléments, le G dans la structure de la phase φ considérée.
Le second terme, , correspond à une énergie de Gibbs de mélange idéal (ou mécanique), dans lequel les constituants se répartissent aléatoirement. L’entropie configurationnelle qui en résulte est décrite par la formule dite de Bragg-Williams
. Cette contribution
est donc connue analytiquement et dépend uniquement de la température et de la composition du mélange.
Enfin, le dernier terme, , correspond à l’enthalpie libre d’excès générée par les interactions entre les constituants du mélange. Ces interactions seront décrites par un ou plusieurs paramètres d’interaction suivant le formalisme de Redlich-Kister [5]. On obtient alors:
Les paramètres d’interaction sont décrits par des fonctions (affines généralement) de la température. Il peut être nécessaire d’utiliser plusieurs ordres n de paramètres d’interaction pour décrire correctement la contribution d’excès du mélange.
Si tous les termes d’interaction sont nuls, on retombe sur la description d’une solution idéale. L’énergie de Gibbs de la solution peut être décrite uniquement par la somme du terme de référence et du terme idéal
.
En revanche, si la solution met en jeu des interactions entre les constituants, alors ce terme d’excès est nécessaire. On dit en général qu’un terme 0L non nul correspond à la description d’une solution régulière (un extremum sur la courbe de exG) si tous les termes des ordres supérieurs sont nuls. S’ils ne sont pas nuls, on parle alors de solutions sous-régulières.
Ces paramètres d’interaction peuvent être positifs (tendance des deux constituants à se repousser), nuls (mélange aléatoire), ou négatifs (tendance des deux constituants à s’attirer mutuellement). Les interactions peuvent être de nature enthalpique (termes a) ou entropique (termes b).
Dans cette description, le terme d’interaction n’apporte pas de contribution au Cp (dérivée seconde par rapport à T nulle). La solution suit donc la règle de Neumann-Kopp: l’enthalpie et l’entropie de mélange sont indépendantes de la température.
On peut représenter les différentes contributions refG, idG, exG et finalement l’enthalpie libre du mélange en fonction de la composition.
 Evolution de l’énergie de référence dans le mélange A-B |
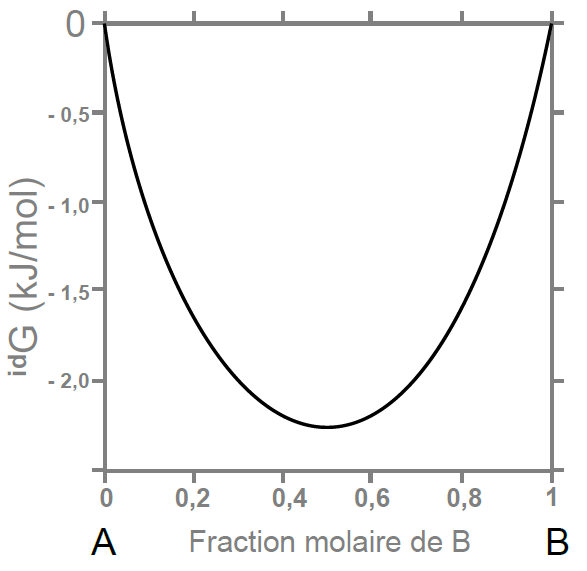 Evolution de la contribution idéale dans le mélange A-B [6] |
 Effet des paramètres d’interaction sur l’énergie d’excès dans le mélange A-B [6] |
 Evolution de l’enthalpie libre du mélange A-B en fonction de la valeur du paramètre d’interaction [6] |
Certains constituants du mélange considéré peuvent ne pas être dans leur état d’agrégation correspondant à l’état stable dans les conditions standard de référence (SER). Pour mieux comprendre cette stabilité de réseau, prenons le cas d’une solution AB de structure cubique à faces centrées (CFC), avec le constituant A cristallisant dans la structure CFC à l’état SER, et le constituant B cristallisant dans la structure cubique centrée (CC) à l’état SER. Pour réaliser cette solution CFC, il est nécessaire de définir tous les constituants à l’état CFC et, par conséquent, il faut définir le constituant B dans l’état CFC, créant une différence d’enthalpie libre entre l’état CFC et CC. C’est ce qu’illustre la figure suivante [6].

Enthalpie libre de référence de la phase CFC : cas où les deux éléments ont des états SER différents [6]
Nous venons de décrire le formalisme utilisé pour traiter le cas des solutions solides de substitution, dites aussi solutions désordonnées, les atomes A et B se substituant sur les mêmes sites du réseau cristallin. Le caractère aléatoire est apporté par la contribution de mélange idéal, et l’écart à cette idéalité est apporté par le terme d’excès.
Les solutions solides ainsi que la phase liquide sont très souvent décrites en utilisant ce formalisme. Il arrive cependant que, dans certaines phases liquides, il existe une mise en ordre à courte distance (SRO, « Short Range Ordering ») autour d’une composition particulière. On peut alors décrire ce phénomène en utilisant d’autres modèles: le modèle associé [7], le modèle ionique [8], le modèle quasi-chimique [9–12]. Certains d’entre eux sont également utilisés pour décrire des solutions solides. Nous vous renvoyons vers les références indiquées pour ces modèles spécifiques, nous ne les détaillerons pas ici.
6. Modèles pour les solutions ordonnées et les composés non-stœchiométriques (modèle en sous-réseaux)
Dans le cas des solutions ordonnées et des composés non-stœchiométriques, il est nécessaire d’avoir recours à des modèles permettant de les décrire dans un contexte cristallographique précis, avec des sites présentant des taux d’occupation différents.
Le modèle à deux sous-réseaux introduit par Hillert et Staffansson [13] a donné naissance au formalisme dit CEF (Compound Energy Formalism), qui sera par la suite étendu pour prendre en charge un grand nombre de sous-réseaux et de constituants dans chaque sous réseau par Sundman et Ågren [14].
Ce modèle en sous-réseaux permet de décrire non seulement des solutions interstitielles, mais aussi des composés avec des domaines d’homogénéité plus ou moins étendus. Chaque sous-réseau est généralement identifié à un ensemble de sites cristallographiques équivalents. Le nombre de ces sous-réseaux est fonction de leur multiplicité et permet de rendre compte de la cristallographie de la phase. Soit la phase décrite par (A,B)m(C,D)n. Les éléments A et B occupent le premier sous-réseau (substitution possible sur le premier sous-réseau), les éléments C et D occupent le second sous réseau (avec possibilité de substitution sur le second sous-réseau). Les multiplicités m et n rendent compte des stœchiométries de chaque sous-réseau. Si un seul élément occupe chacun des sous-réseaux, il s’agit alors d’un cas limite que l’on nomme end-member (qu’on pourrait traduire par composé limite). Ce end-member correspond à une composition ordonnée dans la phase qui peut avoir une réalité physique ou non (stable, métastable, instable). Par permutation, ce modèle en sous-réseaux va définir un jeu de end-members. Dans l’exemple donné ci-dessus, 4 end-members sont générés et devront être décrits par un polynôme représentant leur énergie de Gibbs : AmCn, AmDn, BmCn, et BmDn.
On comprend ici aisément qu’une solution de substitution telle que décrite précédemment constitue un cas particulier de ce modèle, avec un seul sous-réseau et avec pour end-member les deux éléments purs pris dans la structure de la solution. De même, si un seul constituant est présent dans chaque sous-réseau, le modèle revient à celui du composé stœchiométrique.
Dans ce formalisme en sous-réseaux, il est possible de faire apparaître des lacunes (vacancies), ce qui sera très pratique pour décrire certains écarts à la stœchiométrie lorsque c’est ce mécanisme qui est mis en jeu ainsi que les solutions interstielles.
Afin d’utiliser ce type de modèle, il est nécessaire de définir la fraction de sites occupés par l’espèce i dans le sous-réseau s.
Où est le nombre d’atomes du constituant i dans le sous-réseau S, et NS est le nombre total de sites du sous-réseau S.
Cette expression peut être généralisée pour l’intégration de lacunes dans le sous réseau S:
Les fractions molaires de chaque constituant i sont alors données par:
L’enthalpie libre est définie en appliquant le modèle de solution substitutionnelle sur chaque sous-réseau sur lequel il peut y avoir un mélange. Nous retrouvons la somme de l’enthalpie libre de référence, de l’enthalpie libre configurationnelle (idéale), et de l’enthalpie libre d’excès. Si on explicite chacun de ces termes dans le cadre de ce modèle en sous réseaux, il vient:
Avec par exemple (à reproduire pour les autres termes):
Dans ces expressions, correspond à la fraction du sous-réseau occupée par l’espèce i. Les enthalpies libres
sont les énergies des quatre end-members.
Ce formalisme, présenté ici pour deux sous-réseaux, peut être généralisé pour un grand nombre de sous-réseaux et de constituants.
7. Exemple d’illustration
Nous allons regarder de plus près les modèles utilisés pour un système bien connu, le système Fe-C. Il a l’avantage de proposer une description du liquide avec un modèle de substitution et les autres phases solides avec un modèle en sous-réseaux. Ces descriptions proviennent de la modélisation du système par Gustafson [15].
Dans ce système, nous ne prendrons pas en considération les aspects magnétiques par souci de simplification (mais ils existent et doivent être traités).
Voici la liste des phases, avec les modèles qui leur sont associés.
| Phase | Modèle | Type | Param. Interaction |
| Liquide | (C,Fe)1 | Substitution | 0LC,Fe, 1LC,Fe, 2LC,Fe |
| Ferrite (α, δ) | (Fe)1(C,Va)3 | Insertion | 0LFe:C,Va |
| Austénite (γ) | (Fe)1(C,Va)1 | Insertion | 0LFe:C,Va |
| Cémentite | (Fe)3(C)1 | Comp. défini | – |
| Graphite | (C)1 | Comp. défini | – |
La phase liquide utilise un modèle de substitution avec les deux éléments Fe et C dans l’unique sous-réseau. L’écart à l’idéalité du mélange est caractérisé par des paramètres d’interaction. Le nombre de ces paramètres d’interaction est à mettre en parallèle de la forme de la courbe d’enthalpie libre de mélange.
Le Fer α (ferrite) est décrit ici comme une solution solide d’insertion en utilisant un modèle à deux sous-réseaux. On la retrouve à plus haute température sous la dénomination Fer δ, mais la structure est identique. La stœchiométrie des sous-réseaux (1:3) est à corréler avec le nombre de sites octaédriques de la structure cubique centrée (CC) par atome métallique (3). On remarque la présence d’un seul paramètre d’interaction 0LFe:C,Va. Si nous développons les end-members, on trouve (Fe)1(Va)3, qui correspond au Fer α, et (Fe)1(C)3, qui correspond à une structure FeC3 métastable qui ne s’exprimera pas ici.
Le Fer γ (austénite) est lui aussi décrit comme une solution solide d’insertion sur la base d’un modèle à deux sous-réseaux. Les coefficients stœchiométriques sont à rapprocher de la multiplicité des sites, le carbone se dissolvant dans les sites octaédriques (1 site octaédrique par atome métallique). Un seul paramètre d’interaction est utilisé ici pour décrire l’écart à l’idéalité. Si nous développons les end-members, on trouve (Fe)1(Va)1, qui est la description du Fe γ, et (Fe)1(C)1 qui correspond à la description d’une phase FeC de structure NaCl métastable qui ne s’exprimera pas ici, d’autres phases, ou plutôt équilibres, plus stables l’en empêchent.
La phase Cémentite est ici déclarée comme un composé défini. Sa composition correspond à celle fixée par les coefficients stœchiométriques des sous-réseaux. Cette phase est métastable dans le binaire Fe-C et ne pourra s’exprimer dans le diagramme de phases stables.
Le graphite est l’état SER du carbone. Il s’agit d’un corps pur, le carbone est seul dans son sous-réseau.
Bien entendu, ce qui a été proposé ici dans le cadre du système binaire Fe-C peut être étendu à des systèmes d’ordres supérieurs (ternaires, quaternaires, quinaires…): c’est une des grandes forces de la méthode CALPHAD!
8. Stratégie de modélisation
Avant d’affiner les différentes variables de nos polynômes de G, il faut faire certainement le travail le plus important: la compilation des données d’entrée (thermodynamiques, de diagramme de phases, ab initio), et faire une sélection critique de ces données. On néglige souvent l’incertitude sur les données, mais c’est pourtant un critère clé non seulement pour la sélection des données à prendre en compte dans l’étape d’optimisation, mais aussi dans la déclaration de ces données dans le moteur d’optimisation (ce qui laissera dans un certain sens une « liberté » au modèle d’ajustement d’évoluer entre des bornes définies par les incertitudes).
Après ce travail de compilation critique des données d’entrée vient le choix du modèle propre à chaque phase. Ce choix doit être fait pour rendre compte de la cristallographie de la phase, mais aussi pour rendre compte correctement de toutes les fonctions de mélange. Ici vont intervenir des considérations physiques et chimiques des solutions: interactions entre constituants, mise en ordre autour d’une ou plusieurs compositions particulières, démixtion…
Ces deux étapes clés réalisées, la modélisation à proprement parler peut commencer, pendant laquelle toutes les variables vont être affinées successivement, phase par phase, à partir des données expérimentales et théoriques, et à partir des modèles choisis.
La figure ci-dessous, très largement inspirée de celle Lukas [3], donne une idée du processus de modélisation.

Description des étapes de modélisation : analyse critique des données et étapes d’optimisation
En sortie, on obtient un jeu de variables ajustées pour les polynômes de G (descriptions optimisées sur la figure) décrivant l’ensemble des phases du système. Cet ensemble de polynômes constitue la base de données du système permettant de réaliser des calculs d’équilibre: tracé de fonctions thermodynamiques, évolution d’une propriété, et bien entendu retracer le diagramme de phases.
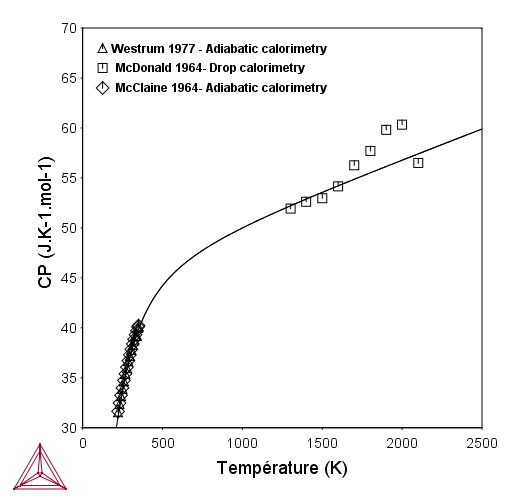 Evolution du Cp de la phase HfC en fonction de la température |
 Incréments enthalpiques de la phase HfC |
 Evolution de l’enthalpie de formation de la phase HfC en fonction de sa composition |
 Diagramme de phases du système Hf-C |
Dans cet exemple, nous allons illustrer la modélisation de la phase HfC1-x, sous-stœchiométrique en carbone. Après compilation des données de la littérature (nous n’avons pas tout représenté ici), il faut choisir le modèle permettant de décrire la phase HfC. Le choix s’est porté sur le modèle en sous-réseaux suivant: (Hf)1(C,Va)1. La structure de la phase est de type NaCl, les atomes de carbone occupant les sites octaédriques de la structure CFC de Hf, et ce modèle génère deux end-members, l’un correspondant à HfC stœchiométrique type NaCl et l’autre à Hf CFC. L’état SER pour Hf correspond à la structure HC (hexagonale compacte). Il faudra donc considérer l’état Hf CFC par rapport à l’état SER (HC) avec une énergie de stabilité de réseau. On utilisera donc directement cette description de Hf CFC en important le polynôme correspondant depuis la base PURE. On peut noter que c’est exactement le même modèle qu’utilisé précédemment pour l’austénite. La différence tient au fait que dans Fe-C, c’est le fer CFC qui est stable et le carbure de type NaCl qui est métastable, alors que dans Hf-C, c’est la carbure NaCl qui est stable et le Hf CFC qui est métastable.
Voici un exemple du polynôme utilisé pour le end-member correspondant à HfC:
Les variables à affiner sont notées de V1 à V6.
La première étape consiste à affiner les variables liées au Cp et à l’évolution de l’enthalpie en fonction de la température (heat content) (V3 à V6). On compare notre affinement avec les données de la littérature en traçant l’évolution du Cp et de l’enthalpie en fonction de la température avec les données expérimentales en surimpression. Si l’affinement parait convenable, on fixe ces variables. Sinon, il faut revoir la sélection des données. Parfois il peut être nécessaire de modifier le polynôme lui-même, mais nous n’aborderons pas ce cas ici.
Nous avons des valeurs d’enthalpie de formation à notre disposition, et nous nous en servons pour affiner V1. Il est aisé de comparer les valeurs expérimentales avec la valeur d’enthalpie de formation calculée après l’affinement de V1. Si des écarts subsistent, notamment au-delà de l’incertitude renseignée, il sera nécessaire de revoir les données d’entrée, soit en donnant des poids différents à chaque entrée, soit en éliminant certaines données expérimentales sur la base d’un raisonnement critique.
Si la phase liquide a été préalablement modélisée, nous pourrons utiliser les données liées à la congruence (composition et température et de la congruence ) pour fixer la contribution entropique V2. On vérifie après affinement que la fusion a bien lieu à la température attendue, sinon il faudra revoir les données d’entrée (réduire la dispersion, appliquer des poids différents pour certaines données).
Deux paramètres d’interaction (ordre 0 et ordre 1) sont nécessaires pour décrire l’étendue de composition de la solution solide HfC1-x:
Dans notre cas, les valeurs d’enthalpie de formation en fonction de la teneur en carbone dans la phase, les données issues des équilibres biphasés (conodes), ainsi que des valeurs d’activité en carbone (non représentées ici) nous permettent de fixer V11, V12, V21 et V22.
Voici comment sont définis les modèles pour les différentes phases du système:
| Phase | Modèle | Param. Interaction |
| Liquide | (Hf,C)1 | 0L, 1L, 2L, 3L |
| CFC | (Hf)1(C,Va)1 | 0L, 1L |
| CC | (Hf)1(C,Va)3 | 0L |
| HC | (Hf)1(C,Va)0,5 | 0L, 1L, 2L |
Il est important de toujours retracer les fonctions thermodynamiques de toutes les phases pour vérifier la cohérence des affinements avec les données de la littérature tout au long du processus de modélisation. Bien souvent l’étape ultime est le tracé du diagramme de phases sur la base des données affinées par le processus de modélisation, mais il ne doit pas se substituer à la vérification minutieuse des fonctions thermodynamiques!
9. Description des données dans le formalisme 3ème génération
Comme évoqué plus haut, cette nouvelle génération de formalisme permettra de mieux interfacer la méthode CALPHAD et les calculs de premier principe comme la DFT, notamment par la prise en compte des données dès 0 K, ce qui permettra de réconcilier CALPHAD et troisième principe de la thermodynamique.
Cette troisième génération de base de données est dédiée à la description des corps purs dès 0 K, tous les autres constituants (composés et solutions) pourront alors être décrits sur les mêmes principes que ceux présentés dans la partie CALPHAD.
L’idée ici est d’utiliser le modèle d’Einstein et un polynôme, permettant d’avoir à 0 K Cp = S = 0. Cette description est donnée par [16]:
pour 0K < T < Tf
Avec Tf : la température de fusion, E0 : l’énergie à 0 K, θE : la température d’Einstein, a et b : les corrections électroniques et anharmoniques.
pour Tf < T < 6000K
Avec a’, b’, c’: des variables de contrainte (égalité des deux expressions notamment sur le Cp à Tf ainsi que pour sa première dérivée), H’ et S’: enthalpie et entropie de fusion de la phase solide. On suppose de surcroît que cette expression donne une valeur de Cp, pour le solide, égale à celle du liquide bien au-delà de la température de fusion.
Il est possible d’utiliser un modèle spécifique pour décrire le liquide bien en dessous de sa température de solidification, que l’on appelle « two-state model » [17,18].
Avec où
et
représentent les énergies de Gibbs du constituant A pour lequel les atomes sont dans un état liquide ou amorphe.
A basse température, , donc
tend vers 0 et
Avec la fonction d’Einstein,
la température d’Einstein correspondant à cet état amorphe, a et b sont des constantes décrivant la stabilité de cet état amorphe définies à partir de la température et de l’enthalpie de fusion.
A plus haute température, l’exponentielle précédente devient >>> 1, et dans ce cas:
Où est un polynôme de la température:
Les variables c, d, e, …, sont obtenues à partir des propriétés du liquide et de la température de transition entre la forme amorphe et liquide.
Pour aller plus loin sur le développement des bases de données de troisième génération, un bon point de départ est certainement ces articles issus des précédents Ringberg workshops [17,19–22].
10. La DFT au service de la méthode CALPHAD
L’usage de la DFT à destination de la méthode CALPHAD [23,24] s’est développé dans un premier temps pour atteindre des données impossibles à déterminer expérimentalement, soit à cause des limites des appareillages, soit parce que les phases concernées sont métastables. De plus, l’enthalpie de formation n’est pas une information facile à obtenir par les méthodes expérimentales, alors qu’elle est « presque » directe par les méthodes de premier principe. Les choses se corsent pour l’obtention du Cp, issu de calculs de phonons, souvent longs, et tributaires de la taille de cellule adoptée (dépendante de la cristallographie, des taux d’occupation des sites, des lacunes) pour décrire la phase étudiée. Dans le cas de l’étude de solutions par les méthodes ab initio, il faut également recourir à des méthodes plus complexes (SQS: Special Quasirandom Structures [25], CEM: Cluster Expansion Method [26] et CVM: Cluster Variation Method [27,28]). Des outils comme la compilation ATAT (Alloy Theoretic Automated Toolkit) [24] permettent de prendre en charge un partie de ces calculs, et de produire en sortie une base de données utilisable par des logiciels comme Thermo-Calc. Pour illustrer ces liens entre calculs ab initio (DFT, CVM, Monte Carlo) et CALPHAD, la figure suivante est un excellent exemple de ce qui est possible de réaliser [29].
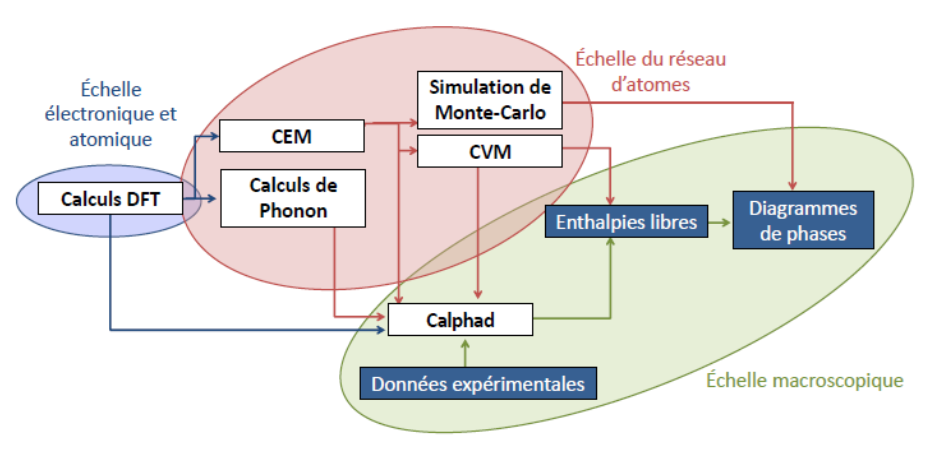
Articulation des méthodes de modélisation, de l’échelle atomique à l’échelle macroscopique.
11. Pour aller plus loin
Pour ceux qui auraient besoin de plus d’informations que celles présentées ici, n’oubliez pas que le GdR organise des écoles d’été, et que de nombreuses autres formations sont proposées (SATA : School for Advanced Thermodynamic Assessments, dont la dernière édition a eu lieu en 2017, et d’autres formations plus locales dans le cadre de Masters). Les journées annuelles du GdR sont également un excellent moyen d’échanger avec des spécialistes du domaine et pour vous de présenter vos problématiques. N’oubliez pas que l’important dans toute modélisation reste la qualité des données sur lesquelles elle s’appuie.
[1] P.J. Spencer, A brief history of CALPHAD, Calphad. 32 (2008) 1–8. https://doi.org/10.1016/j.calphad.2007.10.001.
[2] N. Saunders, A.P. Miodownik, CALPHAD (Calculation of Phase Diagrams): A Comprehensive Guide, Elsevier, 1998.
[3] H. Lukas, S.G. Fries, B. Sundman, Computational Thermodynamics: The Calphad Method, Cambridge University Press, 2007.
[4] A.T. Dinsdale, SGTE data for pure elements, Calphad. 15 (1991) 317–425. https://doi.org/10.1016/0364-5916(91)90030-N.
[5] O. Redlich, A.T. Kister, Algebraic Representation of Thermodynamic Properties and the Classification of Solutions, Ind. Eng. Chem. 40 (1948) 345–348. https://doi.org/10.1021/ie50458a036.
[6] J. Jourdan, Étude expérimentale et thermodynamique des systèmes erbium-oxygène-zirconium et gadolinium-oxygène-zirconium, These de doctorat, Paris Est, 2009. http://theses.fr/2009PEST0035.
[7] F. Sommer, Association Model for the Description of the Thermodynamic Functions of Liquid Alloys. I.–Basic Concepts, Zeitschrift Fur Metallkunde. 73 (1982) 72–76.
[8] M. Hillert, B. Jansson, B. Sundman, J. ågren, A two-sublattice model for molten solutions with different tendency for ionization, MTA. 16 (1985) 261–266. https://doi.org/10.1007/BF02815307.
[9] A.D. Pelton, P. Chartrand, The modified quasi-chemical model: Part II. Multicomponent solutions, Metall and Mat Trans A. 32 (2001) 1355–1360. https://doi.org/10.1007/s11661-001-0226-3.
[10] A.D. Pelton, S.A. Degterov, G. Eriksson, C. Robelin, Y. Dessureault, The modified quasichemical model I—Binary solutions, Metall and Materi Trans B. 31 (2000) 651–659. https://doi.org/10.1007/s11663-000-0103-2.
[11] P. Chartrand, A.D. Pelton, The modified quasi-chemical model: Part III. Two sublattices, Metall and Mat Trans A. 32 (2001) 1397–1407. https://doi.org/10.1007/s11661-001-0229-0.
[12] A.D. Pelton, P. Chartrand, G. Eriksson, The modified quasi-chemical model: Part IV. Two-sublattice quadruplet approximation, Metall and Mat Trans A. 32 (2001) 1409–1416. https://doi.org/10.1007/s11661-001-0230-7.
[13] M. Hillert, L.-I. Staffansson, The Regular Solution Model for Stoichiometric Phases and Ionic Melts., Acta Chem. Scand. 24 (1970) 3618–3626. https://doi.org/10.3891/acta.chem.scand.24-3618.
[14] B. Sundman, J. Ågren, A regular solution model for phases with several components and sublattices, suitable for computer applications, Journal of Physics and Chemistry of Solids. 42 (1981) 297–301. https://doi.org/10.1016/0022-3697(81)90144-X.
[15] P. Gustafson, Thermodynamic evaluation of the Fe-C system, Scandinavian Journal of Metallurgy. 14 (1985) 259–267.
[16] Q. Chen, B. Sundman, Modeling of thermodynamic properties for Bcc, Fcc, liquid, and amorphous iron, JPE. 22 (2001) 631–644. https://doi.org/10.1007/s11669-001-0027-9.
[17] J. Agren, B. Cheynet, M.T. Clavaguera-Mora, K. Hack, J. Hertz, J. Sommer, U. Kattner, Workshop on thermodynamic models and data for pure elements and other endmembers of solutions: Schloβ Ringberg, Febr. 21, to March 3, 1995, Calphad. 19 (1995) 449–480. https://doi.org/10.1016/0364-5916(96)00003-X.
[18] J. Agren, Thermodynamics of Supercooled Liquids and their Glass Transition, Physics and Chemistry of Liquids. 18 (1988) 123–139. https://doi.org/10.1080/00319108808078586.
[19] B. Sundman, F. Aldinger, The Ringberg workshop 1995 on unary data for elements and other end-members of solutions, Calphad. 19 (1995) 433–436. https://doi.org/10.1016/0364-5916(96)00001-6.
[20] B. Sundman, H.-J. Seifert, F. Aldinger, The Ringberg Workshop 1996 on solution modelling, Calphad. 21 (1997) 139–141. https://doi.org/10.1016/S0364-5916(97)00018-7.
[21] M. Zinkevich, F. Aldinger, B. Sundman, The Ringberg workshop 2005 on Thermodynamic Modeling and First-Principles Calculations, Calphad. 31 (2007) 2–3. https://doi.org/10.1016/j.calphad.2006.02.003.
[22] M. Palumbo, B. Burton, A. Costa e Silva, B. Fultz, B. Grabowski, G. Grimvall, B. Hallstedt, O. Hellman, B. Lindahl, A. Schneider, P.E.A. Turchi, W. Xiong, Thermodynamic modelling of crystalline unary phases, Physica Status Solidi (b). 251 (2014) 14–32. https://doi.org/10.1002/pssb.201350133.
[23] Z.-K. Liu, First-Principles Calculations and CALPHAD Modeling of Thermodynamics, J. Phase Equilib. Diffus. 30 (2009) 517. https://doi.org/10.1007/s11669-009-9570-6.
[24] A. van de Walle, R. Sun, Q.-J. Hong, S. Kadkhodaei, Software tools for high-throughput CALPHAD from first-principles data, Calphad. 58 (2017) 70–81. https://doi.org/10.1016/j.calphad.2017.05.005.
[25] A. Zunger, S.-H. Wei, L.G. Ferreira, J.E. Bernard, Special quasirandom structures, Phys. Rev. Lett. 65 (1990) 353–356. https://doi.org/10.1103/PhysRevLett.65.353.
[26] J.M. Sanchez, F. Ducastelle, D. Gratias, Generalized cluster description of multicomponent systems, Physica A: Statistical Mechanics and Its Applications. 128 (1984) 334–350.
[27] J.L. Morán-López, J.M. Sánchez, Theory and applications of the cluster variation and path probability methods, Springer Science & Business Media, 2012.
[28] R. Kikuchi, A Theory of Cooperative Phenomena, Phys. Rev. 81 (1951) 988–1003. https://doi.org/10.1103/PhysRev.81.988.
[29] N. Bourgeois, Modelling of metal-hydrogen systems by DFT, CVM and Calphad method, Thèse de doctorat, Université Paris-Est, 2017. https://tel.archives-ouvertes.fr/tel-01730627.